
IGV 是常用的基因组浏览工具之一,但在项目交付与论文级出图中,其在批量制图、统一风格和自动化方面存在一定局限。
pyGenomeTracks 采用命令行与配置文件驱动,更适合:
l多样本统一风格对比
l批量输出指定区域图片
l高分辨率论文与报告配图
因此,pyGenomeTracks 常被作为 IGV 的有效补充或替代方案。
在一些生信分析项目中,将测序信号直观地展示在基因组上,是用户最容易理解、有说服力的结果形式之一。
本文将系统介绍 pyGenomeTracks 常用的软件命令及关键参数,帮助读者理解基因组可视化图像是如何生成的。
一、为什么要做基因组信号可视化?
在各类基于测序的基因组学实验中(如 ChIP-seq、ATAC-seq、DAP-seq 等),我们的核心目标,都是在全基因组尺度上观察某个因子或染色质特征“出现在哪里、强不强"。
如果只用表格或一串数值来展示结果,这些信息往往显得抽象而零散:
峰值有多少、富集倍数是多少,看起来“很科学",但并不直观,也不利于快速判断实验到底做得好不好。
而基因组信号可视化,正是把这些数据“画到染色体上"。
通过将测序信号转化为沿染色体连续变化的曲线,我们可以一眼看到:
l目标因子是否在启动子、增强子等特定区域明显富集
l不同重复样本之间的信号形态是否一致、稳定
l实验样本与 Input 或背景对照之间,是否存在清晰可辨的差异
这种“看图说话"的方式,不仅能帮助我们快速评估实验质量,也为后续的峰值筛选、功能注释和生物学解读提供了直观可靠的依据。
二、本案例使用的软件工具
本流程主要使用 pyGenomeTracks 软件包中的两个工具:
1. make_tracks_file:用于根据已有结果文件,自动生成可视化所需的配置文件。
2. pyGenomeTracks:用于读取配置文件,在指定的基因组区域内绘制信号图像。
三、软件获取与安装
推荐使用 Conda 安装 pyGenomeTracks:
conda install -c bioconda pygenometracks -y
安装完成后可通过以下命令检查:
lpyGenomeTracks --help
lmake_tracks_file --help
四、输入数据文件说明
1.BigWig 文件(信号文件)
BigWig 文件用于存储测序 reads 在基因组各位置的覆盖强度或归一化信号值,是绘制基因组信号曲线的基础数据形式。
相比原始的 BAM 文件,BigWig 体积更小、读取速度更快,尤其适合用于基因组浏览器或绘图工具进行连续信号展示。
在本案例中,BigWig 文件包括:
lS1.bw:实验样本重复 1 的基因组信号
lS2.bw:实验样本重复 2 的基因组信号
lInput.bw:Input 对照样本
用于评估背景噪音和非特异性信号通过同时展示多个 BigWig 文件,可以直观比较不同重复之间的一致性,以及实验样本与对照之间的差异。
2.BED 文件(区域文件)
BED 文件用于标记基因组上的离散功能区域,例如 peak、结合位点或候选调控区域。
与 BigWig 所展示的连续信号不同,BED 文件更关注“哪里发生了显著事件"。
在本案例中,使用 peak_location.bed 文件来展示预测得到的转录因子结合区域。
将 BED 区域叠加到信号轨迹上,可以帮助我们判断:
lpeak 是否对应真实、稳定的信号
l富集信号是否集中于少数明确的区域,还是分散且噪音较大
l不同样本中同一peak区域的信号表现是否一致
3.GTF 文件(注释文件)
GTF 文件用于提供基因组结构注释信息,包括基因、转录本、外显子、内含子以及启动子等功能单元的位置。在基因组信号可视化中,GTF 文件的作用主要体现在两个方面:
l为信号和 peak 提供生物学背景,帮助判断其位于基因的哪个结构区域
l解读信号的潜在功能意义,例如是否偏向启动子区域或基因间区
通过将 GTF 注释与 BigWig 信号、BED 区域同时展示,可以实现从“信号存在"到“信号有什么生物学含义"的过渡。
五、自动生成 tracks.ini 配置文件(软件命令)
在实际分析中,不需要手工编写复杂的配置文件,而是通过以下命令自动生成:make_tracks_file --trackFiles S1.bw S2.bw Input.bw peak_location.bed sample.gff3 --out tracks.ini
该步骤的作用可以理解为:告诉软件需要展示哪些文件,以及每个文件在图中作为一条独立轨道显示。
生成的 tracks.ini 文件中已经包含了轨道名称、颜色、高度等基础设置。
六、统一信号显示范围(常用参数说明)
在默认情况下,每个 BigWig 信号轨道会根据自身数据自动缩放显示范围。在多样本对比时,这种方式容易造成视觉上的误解。因此,通常会人为设定统一的信号显示上限。
七、绘制基因组信号图(核心命令)
完成配置文件修改后,即可使用以下命令绘制基因组信号图:
pyGenomeTracks --tracks tracks.ini --region NC_083379.1:10813150-10833150 -o image.png --dpi 300
参数解释:
ltracks:指定轨道配置文件。
lregion:指定绘制的基因组区域,格式为“染色体:起始位置-终止位置"。
lo:输出图片文件名。
ldpi:输出图片分辨率,300 dpi 适合用于项目报告和论文。
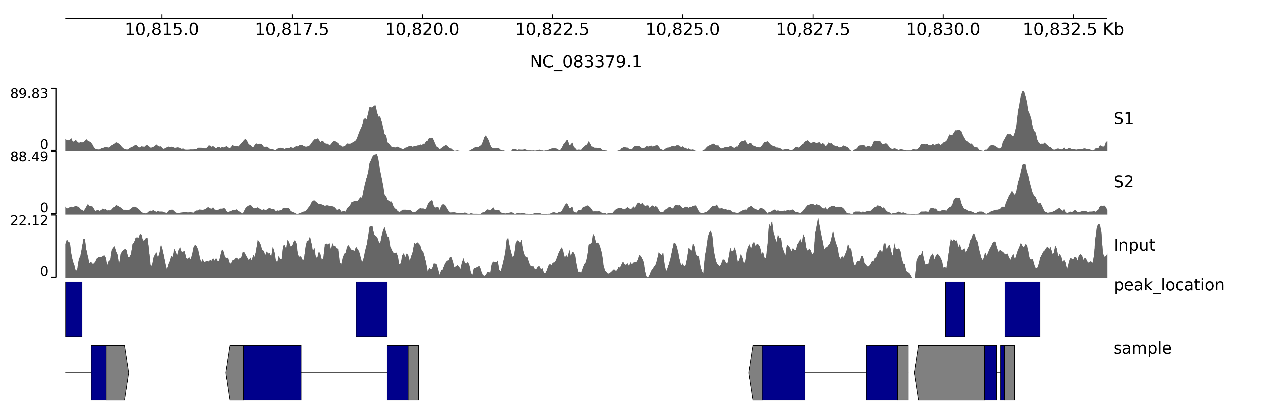
图 1 默认配置示例图
八、tracks.ini 中常关注的参数
在配置文件中,以下参数对图像理解重要:
ltitle:轨道名称,用于区分不同样本
lheight:轨道高度,避免图像过于拥挤
lcolor:轨道显示颜色这些参数直接影响图像的清晰度和可读性。
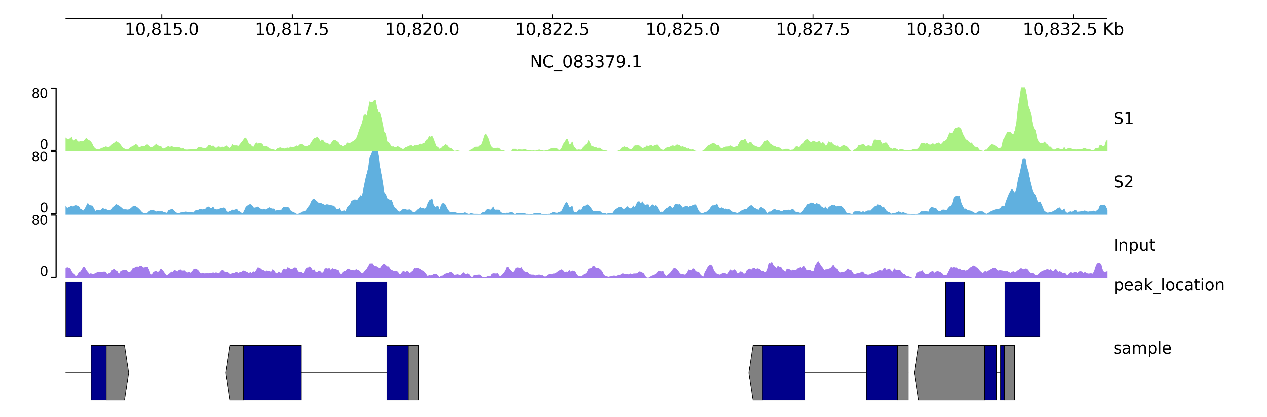
图 2 修改配置文件示例图
九、可以从结果图中获得哪些信息?
通过最终生成的基因组信号图,用户可以直观看到:
l目标区域是否存在明显富集
l不同重复样本之间的信号一致性
lInput 和 IGG 等是否呈现低背景
lpeak 是否与高信号区域对应
这些都是判断实验是否成功和结果是否可靠的重要依据。
附录:pyGenomeTracks 可视化配置参数完整对照表
(一)坐标轴轨道(x-axis)
.png")
(二)空白间隔轨道(spacer)
.png")
(三)连续信号轨道(BigWig)
1. 基本信息
1.png")
2. 颜色与透明度
2.png")
3. Y 轴范围与分辨率
3.png")
4. 缺失值与统计方式
4.png")
5. 信号绘制方式
5.png")
6. 信号运算(实时计算)
6.png")
7. 数据变换
7.png")
8. 文件类型
8.png")
(四)功能区间轨道(BED)
1. 基本信息
1.png")
2. 颜色与边框
2.png")
3. 显示方式
3.png")
4. 标签与字体
4.png")
5. 箭头与结构参数
5.png")
6. 文件类型
6.png")
(五)基因注释轨道(GTF)
1. 基本信息
1.png")
2. 基因名称与转录本处理
2.png")
3. 颜色与显示方式
3.png")
4. 行数、标签与字体
4.png")
5. 箭头与结构参数
5.png")
6. 文件类型
6.png")

